Gene flow in mainland vs. island populations of Anopheles gambiae
Connectivity of populations on the genetic level, resulting in gene flow across long distances, is an important factor for understanding evolutionary processes on the meta-population level. In mosquitoes it is also highly relevant to disease control efforts because it allows e.g. predicting the spread of resistance alleles and monitoring engineered mosquitoes that have been released for malaria control. In this project we compare the degree of connectivity/isolation to neighboring and distant populations of two African populations of the malaria vector Anopheles gambiae on the genomic level. One of the focal populations is from Mali and represents a population that presumably has a large effective population size and is highly connected to neighboring populations. The other focal population is from an archipelago consisting of several islands of different size, located at Africa’s east coast between Mozambique and Madagascar. This population presumably features more intra- and inter-population structure due to the geographically patchy and isolated habitat.
We sampled mosquitoes from Mali, the Comoros and several places in between and produced individual whole genome re-sequencing data for more than a hundred specimens. We analyze the whole dataset for signs of historical shifts in effective population size, cross-coalescence, genetic isolation and admixture. This will produce unprecedented insight into the interdependence of geographical features and genetic patterns on a broad scale level. We will carefully evaluate potential field sites for release experiments with genetically engineered mosquitoes based on these findings.
This project was completed as part of the University of California Irvine Malaria Initiative.
Identifying immune genes responsible for susceptibility to Plasmodium parasites in A. gambiae in Mali and Cameroon
Significant data implicate various signaling pathways in the mosquito immune system in the regulation of malaria parasite development in the Anopheles gambiae midgut. However, no data are available to confirm that these pathways regulate parasite development in nature. The Vector Genetics lab, in collaboration with the laboratory of Dr. Shirley Luckhart in the Department of Medical Microbiology and Immunology, School of Medicine at UC Davis, have chosen to address this issue using a SNP association approach with field-collected mosquitoes from Mali and Cameroon, countries with high malaria transmission that include the geographic distribution of the most genetically diverged A. gambiae populations yet described.
The first component of this project includes population genetic analyses. Plasmodium falciparum infected and uninfected A. gambiae are being collected from sites in Mali and Cameroon. These sites were carefully selected to include all of the mosquito genetic diversity known to exist in the region (three molecular forms and chromosome inversion polymorphism). Mosquito samples are grouped by site with respect to infection, molecular form and karyotype. Each mosquito is then genotyped for roughly 384 immune signaling gene SNPs and examined if those SNPs are correlated with infection. If any of the 384 SNPs occurs more frequently in infected versus uninfected mosquitoes, they may contribute to making the mosquito susceptible to Plasmodium infection.
In the second project component, selected SNPs of interest will be analyzed to determine their effects on mosquito protein function and on susceptibility to P. falciparum infection in the laboratory using both mosquito cell lines and live mosquitoes from colony. Two genetically distinct strains of A. gambiae, which have been genotyped for the SNPs of interest will be used in the laboratory studies. We will use inhibitors and DNA transfection protocols to mimic the effects of SNP-containing alleles on P. falciparum development in artificially infected mosquitoes. This work will take functional data from Dr. Luckhart’s lab and from the labs of our colleagues to examine the importance of the selected immune signaling pathways in field-collected mosquitoes. These studies will facilitate selection of appropriate gene targets for strategies involving genetically modified mosquitoes for malaria control and provide critical new insights into the immunity in A. gambiaepopulations.
This project was conducted in collaboration with Dr. Sékou Traoré, Malaria Research and Training Center, University of Bamako, Mali and Dr. Etienne Fondjo, National Malaria Control Programme, Youndé, Cameroon.
Ecological and genetic determinants of malaria transmitting behaviors in Anopheles arabiensis in Tanzania
Anopheles gambiae is frequently referred to as the most important vector of malaria in Africa and has been the main focus of malaria vector research. Despite this attention, there is growing evidence that it is not this species, but its sister species A. arabiensis that is increasingly responsible for malaria transmission in Africa. Reports indicate that in areas of high insecticide treated net (ITN) coverage, A. arabiensis outcompetes An. gambiae s.s and has become the dominant vector species in many locations. If this phenomenon continues as large-scale ITN programs are rolled out across Africa, this species could become the only medically relevant vector in many parts of the continent. Consequently the ecology, vectorial competence and population genetics of this somewhat neglected vector merit particular attention in preparation for future vector control scenarios.
This research program integrates vector population genomics, ecology and vector behavior with the goal of understanding the determinants of two mosquito behavioral phenotypes crucial to the transmission and control of malaria: (1) host preference and (2) adult resting behavior. Our approach builds upon a sizeable base of preliminary work, conducted in our laboratory, which has identified an extensive panel of A. arabiensis SNP markers, and preliminary field work in Tanzania that has identified a range of appropriate sites where sampling methods have been piloted and the behavior of A. arabiensis is known to vary.
A. arabiensis mosquitoes will be intensively collected from four villages in the Kilombero Valley of Tanzania during the wet and dry seasons to determine the association between their feeding and resting phenotype and environmental factors that vary temporally and spatially (component #1). DNA will be extracted from individual samples and multi-locus SNP genotypes determined from each individual. Genotypes will be organized by phenotype (exophilic vs. endophilic and human fed vs. animal fed) and analyzed to determine SNP allele associations with each phenotype after correcting for population structure (component #2).
Knowledge of the genetic basis of these behavioral changes will be vital for prediction of both possible downstream evolutionary responses to current vector control strategies, and also for the development of novel control strategies that improve the application of currently available vector control methods and/or that are based on vector genetic manipulation.
This project was conducted in collaboration with Drs. Heather Ferguson and Daniel Haydon, University of Glasgow, UK, Dr. Gerry Killeen, Ifakara Health Institute, Tanzania and Dr. Eleazar Eskin, University of California, Los Angeles.
The Ecology and Genetics of Anopheles gambiae on Islands
Progress toward the development of genetically modified mosquito (GMM) strains to be used in a genetic control program for mosquito vectors of malaria has reached a point where most of the scientific community involved in this research are beginning to think about field trials. The following recommendation was made by a group of experts in the field of vector biology participating in a meeting entitled “Genetically Engineered Arthropod Vectors of Human Infectious Diseases”: “The exciting progress in molecular strategies for blocking parasite development in vectors has far outpaced the related and essential work on field sites and vector populations that must precede any GM release field trial.” (Imperial College, London 14-15 September, 2001).
We believe that the most promising sites for GMM field trial sites would be on islands off the coast of sub- Saharan Africa. In this project we are conducting studies aimed at assessing the suitability of two island groups. Based on a critical analysis of available data we chose to study A. gambiae populations on islands in the Bijago Archipelago off the coast of Guinea Bissau in West Africa and the Comoros Islands located between Madagascar and Mozambique in East Africa. The project includes:
- collection of basic ecological and biological data for An. gambiae at each site, including relative population densities and adult distribution;
- an assessment of the degree to which island populations are genetically isolated from the nearest mainland populations and from populations on neighboring islands, and a description of the genetic structure of populations on each of the two target islands;
- creation of a GIS system that describes the ecology of each site and is fully integrated with information from the ecology and population genetics studies.
Anopheles gambiae Population Genetics

Malaria is one of the most important infectious diseases of humans with more than 300 million clinical cases and 2-3 million deaths per year, most of these in Africa. Anopheles gambiae is one of the most potent malaria vectors known. This species has a broad geographic distribution, occurring throughout sub-Saharan Africa.

An. gambiae has three pairs of chromosomes, including two autosomes and X/Y sex chromosomes. Like Drosophila the chromosomes in some tissues exist as large polytene chromosomes with distinctive patterns of bands. Cytogenetic studies based on microscopic examination of the polytene chromosomes have revealed extensive polymorphism in the form of chromosome inversions. Earlier work describing the distribution of inversions suggested the existence of subpopulations, identified as “chromosomal forms,” among which gene flow is severely restricted.
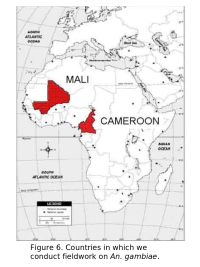
Our research on populations of An. gambiae in Africa is centered on understanding their complex genetic structure. This work is conducted both at field sites in Africa and in our lab located on the 6th floor of Storer Hall at UC Davis. Our fieldwork in Africa includes sites in both Mali and Cameroon.
Using microsatellite DNA markers, we have been able to show that levels of divergence (gene flow) among the chromosomal forms are not uniformly distributed over the genome, being significantly higher at loci within inversions compared to elsewhere in the genome. These results are important because (1) they suggest selection on loci contained within inversions and (2) that subpopulations may not be maintained by assortative mating alone.
By analyzing DNA from sperm recovered from mated females collected in the field we were able to demonstrate strong assortative mating with respect to chromosomal form, but that reproductive isolation is far from complete.
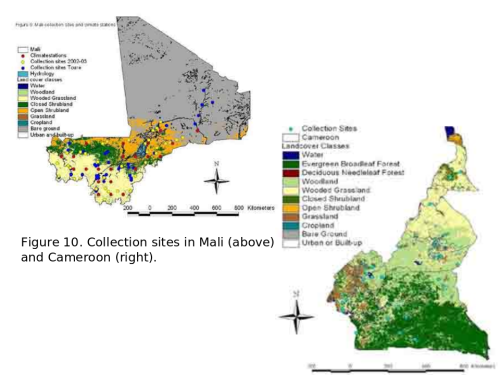
Our current efforts include the development of methods for mapping the spatial boundaries of An. gambiae demes in Mali and Cameroon by incorporating genetic data into GIS maps (in collaboration with Dr. Charles Taylor, Department of Ecology and Evolutionary Biology and Dr. Yong-Kang Xue, Department of Geography both at UCLA). This effort involves extensive field work to obtain field collected mosquitoes from many locations in Mali and Cameroon.
Sand Flies and Leishmaniasis

This research project deals with understanding the complex interactions between insect vectors, pathogens and vertebrate hosts. We use sand flies and leishmaniasis as our model system and are focusing on how these interactions may be understood at the molecular level by studying salivary proteins and the genes that encode them.
The saliva of blood feeding insects contains a fascinating array of proteins that have potent anti-hemostatic activities. Hemostasis describes a collection of complex biochemical pathways that act to inhibit blood loss due to trauma. Hemostasis includes blood clotting, coagulation and vasoconstriction. In addition to their role as anti-hemostatic agents many of these substances are immunomodulators, that is they inhibit the host immune system.
We work with one small protein, known as maxadilan (MAX) that occurs in the saliva of the sand fly, Lutzomyia longipalpis.This sand fly is the vector of the parasite, Lesihmania chagasi, causative agent of visceral leishmaniasis in Latin America. MAX is a potent vasodilator (in fact the most potent vasodilator known). In addition, MAX inhibits macrophage function. It has been shown that MAX is important in the pathogenesis of L. chagasi and that animals immunized with MAX are protected from L. chagasi infection. It has therefore been proposed that MAX may be developed as an effective vaccine for leishmaniasis [Science 2001. 293(5532): p1028].
We have conducted a series of experiments in which we have shown that specific anti-MAX antibodies are produced by animals and humans exposed to Lu. longipalpis bitesand that these antibodies neutralize the vasodilatory activity of
MAX. Consequently, flies feeding on animals with MAX antibodies take smaller bloodmealsand lay fewer eggs than those feeding on naive animals. We reasoned that flies in nature have likely evolved some means of avoiding the host immune system.
We have demonstrated that MAX is hypervariable in natural fly populations and that this variation represents antigenic variation that has evolved in response to the challenge imposed by host antibodies. This is the first description of antigenic variation in a bloodfeeding arthropod.
Lutzomyia longipalpis Population Genetics
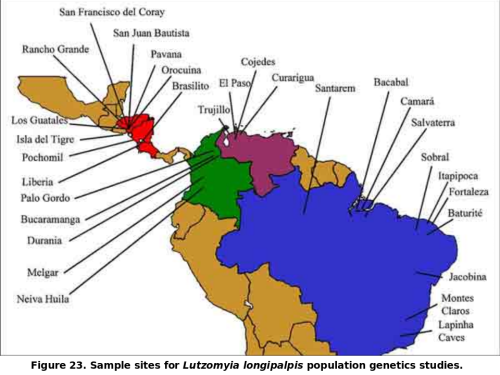
Our work on the population genetics of Lu. longipalpis was conducted in conjunction with the MAX work. We have described the genetic structure of Lu. longipalpis at over 20 sites throughout its range (from northern Argentina to southern Mexico).
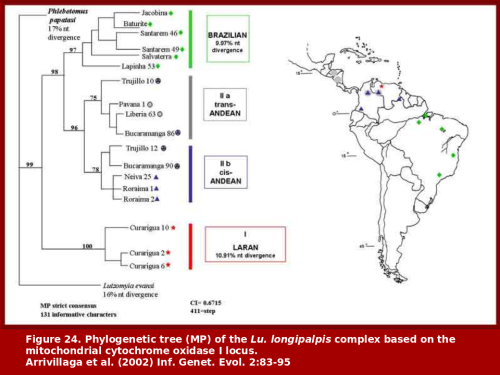
This work was based on isozymes and mitochondrial DNA. We have shown that at least four distinct population groups occur in nature and that these probably represent species. The species status has been confirmed by experimental hybridization, which culminated in the production of sterile hybrid males. In addition, close examination has revealed morphological features that are diagnostic for at least some of our putative species.